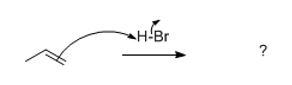
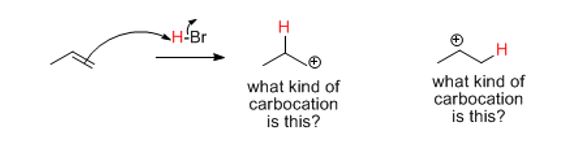
This chapter and more importantly, this class of reaction is known as Electrophilic Addition or simply ‘addition.’ Recall that the double bond above is simply a strong (sigma) bond plus a weak one (pi). Specifically, this class of reaction (that alkenes and alkynes undergo) is named for the rate-determining or slow step of the reaction: Addition of a positively-charged species to the rupturing double bond.
Because the pi-bond is weak and electron-dense (its orbitals are filled and cannot accommodate more) it is attracted to electrophilic or positively (+) charged species)). The alkene (a weak bond constitutes an excess of electron density making it functional) then naturally goes after electrophiles like H+ ions. It wants or "seeks" or "loves" electron scarcity and forms a bond to it. Hence, the name addition.
However, two bonds that are formed don’t add in one step all the time, in all cases, and these reactions often more than one step.
Reactions of Alkenes and Alkynes: Mechanism (AdE3)
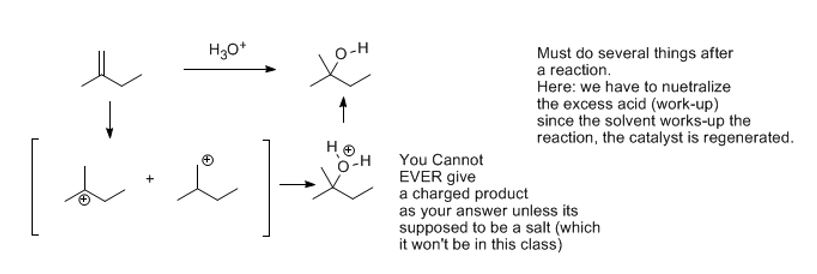
Every reaction of this type for this section deals with the breaking of pi bonds. This creates an important intermediate. This is the class of reactions known as ElectrophilicAddition, the reactions of alkenes and alkynes (C-C pi bonds). The pi-bond is destroyed but where does the pi bond go? To an electrophile leaving a net deficiency on the other carbon.
Since each sp2 carbon has lost a bond, each will gain an atom during the reaction depending on the reagents you use. They will therefore end up sp3 but the reaction as far as mechanism is concerned usually proceeds in two steps.
In the first step, the breaking the pi bond attacks the the nucleophile. An arrow is drawn from the middle of the alkene bond to show:
1) something negatively charged or electron-rich (a double bond) is attacking something electron deficient: therefore, they seek out one another. (+) Plus charges and (-) negative charges attract. The most important thing concerning when drawing a feasible mechanism is the direction the curved arrows (the electrons) go to and where they come from. They go from the source of electron density (something electron-rich) toward a deficit of electron density like a partial or full positive charge.
2) Arrows drawn from the middle of any bond means it is being broken and therefore will not be present in the next drawing. Where the electrons go depends on the barrier to forming a bond. Wherever a more stable entity will form is the basis for the regiochemical outcome.
3) The shaft of the arrow will become a bond in the next drawing or structure. This means you must sure not to violate the octet rule. If something is forming a bonding to a neutral atom, it must lose a bond simultaneously. Avoid any and all violations of the octet rule for the second row elements.
Regiochemistry refers to the specific atoms that selectively react to give a regional or local specificity to the product and is based on the stability of the species formed.
This could be hydrobromic or hydrochloric acid, but even though abbreviations are not helpful, it is important to see that once you’ve learned the reaction, it will be roughly the same experience regardless of the electrophile. It can be any variable much like calculus or algebra.
The first step is attack of the alkene nucleophile onto the electrophile. Using one of the reactions you'll actually use, (since this is how you'll see it):
If one draws out what remains of this sequence of arrows, the next step in the mechanism is ready to be drawn.
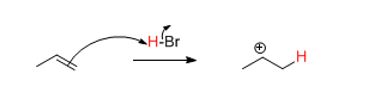
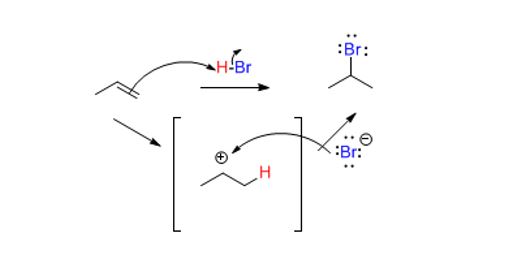
The nucleophile attacks the hydrogen which loses its electrons to the newly formed conjugate base formed. Understanding these elementary steps comes from polarity.
Charges, even partial ones, from polar covalent bonds are sufficient to drive a reaction. Since Br is more electronegative than H, the hydrogen is electron deficient or partially positively charged. If done correctly, a mechanism will show the following intermediate.
The question is where did the electrophile (H) go (what is the regiochemistry?). Since every arrow drawn is a bond in the succeeding structure, when drawing resonance structures or mechanisms, go step by step.
The biggest mistake is to focus on the solution rather than going step by step. It is easiest to make mistakes when you are not drawing and thinking sequentially.
The question: Why does the electrophile go there?
Or stated the other way, why does that carbon attack the electrophilic proton to attach it at the less-substituted (primary) carbon? Why attach to the end of the skeleton?
This was a profound observation made in 1865 by Russian chemist, Vladimir Markovnikov (Romanized spelling: In his notebooks, he signed it as Markownikoff). He noted with the very few tools at his disposal that "in the electrophilic addition of H-X (H-Cl, H-Br, H-I) to asymmetric alkenes…
“the electrophile adds to the carbon having more hydrogens atoms."
—V. Markovniknov
This name is still very important as it now refers to the regiochemistry of the reaction not necessarily the memory device that bears Markovnikov’s name. "The Rich Get Richer" in hydrogens. The carbon with more hydrogens will get more hydrogens. This is so you get a more substituted carbocation (see hyperconjugation).
Notice that on the initial pi or double bond, there is a primary (1o) carbon and a secondary (2o) carbon. These constitute the nucleophile. The reason Markovnikov’s rule works, in most cases, is that the “hole” or “sink” for electrons now appears on a more substituted carbon. That’s why the Br appears with that regiochemistry. Because the choice of one over the other exists, this is a fundamental question. Even Markovnikov however was unaware of the reason behind why his observations were as they were observed.
Markovnikov regiochemistry means when the added group is in place on the backbone of the hydrocarbon. It ends up on more highly-substituted carbon—when there is a choice. If they are equally substituted carbons in the alkene, the reaction occurs at both places and in a predictable ratio of one-to-one (1:1). This is synthetically useless.
When a major product can be formed almost singly is a prudent or favorable reaction. In real life, it would be wasteful to do otherwise. Favoring one product over another is simply a matter of adjusting an experimental parameter such as temperature, pressure, solvent and concentration.
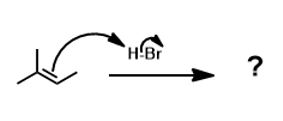
For example, in the following scenario it is possible to indicate which carbocation is formed initially or in higher concentration:
This is an acid-base reaction, as usual. The H atom behaves like a positively charged, or cationic H, or simply a proton (H+). It can attach to either the secondary carbon or the tertiary one. It attaches to the secondary carbon (the secondary carbon has one hydrogen). The other carbon having more substituents (therefore fewer hydrogens, by definition) will ‘get’ the plus charge. That is the major driving force for the formation of the tertiary (3o) carbocation.
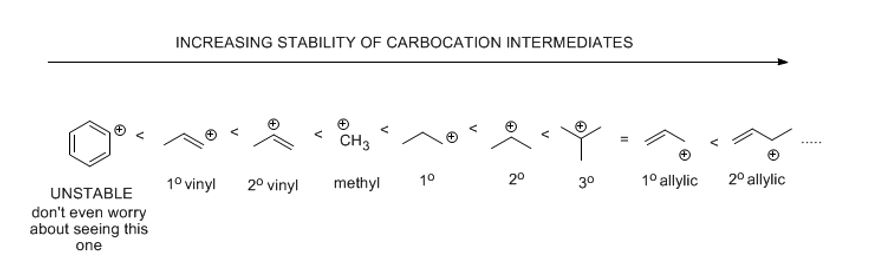
Officially known as carbenium, carbocations are positively charged carbon species. They are the intermediate in this reaction and their stabilities matter. They do not enjoy a full octet of electrons and want to be attacked by a nucleophile to gain an octet. Since the formation of the carbocation is rate limiting, the carbocation must be reasonably stable. The relative stabilities of these is important. The outcome of a reaction depends on whether you can create the intermediate or not. This intermediate (un-isolable) forms most readily because it is tertiary which is more substituted than secondary which is more substituted than primary and so on.
Regiochemistry is not difficult to understand, it's what we are doing right now. Determining where the two atoms or groups that replace both of the carbons in the C-C double bond is the regiochemistry of the reaction. Regio- as in place or positioning, as in the word regional.
The carbon on the end (or terminal carbon) has two hydrogens, the internal sp2 carbon has just one. It's "richer" or more "saturated" with hydrogens. Therefore, the electrophile adds to carbon that has two, not one H. The carbon ends up with three now and the positive charge (the empty p orbital or "hole" in a carbon makes the entire structure a "carbocation".
The stability order of carbocations matters. Even Vladimir didn't know the reason he observed this. He didn't ever know the following facts until quantum mechanics was discovered circa the 1920-30s.
Hyperconjugation.
Just know that the more substituted a carbocationic center is, the more stable it is and the faster it forms. Tertiary is better than secondary which is better than primary which is better than methyl.
Carbocations have different stabilities depending on how many carbons are attached to the positively charged carbon. If there is one carbon attached, it is called primary, if two are attached, it's secondary, and so tertiary (3o) is three. There is no quaternary carbocation (there cannot be an empty orbital on a carbon attached to four carbon groups already.
A more stable carbocation (lower energy) is a stronger possibility and that's what you'll obtain as your intermediate. When there is a choice it is unadvised to make anything less stable than a secondary carbocation. In other words, primary carbocations do not form in a reasonable mechanism up to this point.
Specifically, had you done the following, it would've been wrong. This is the nature of the regiochemistry of a reaction (you don't talk about it much but that is the crux of the problem. How the H and Br are juxtaposed is the most important prediction about the product you can make.
You must be able to distinguish immediately whether a carbon is primary, secondary, or tertiary and consider the stability order of these carbocations.
The carbocation intermediate occupies a position in the mechanism. It corresponds to where the energy reaches a minimum. Draw the product after the first arrow (that's the overall reaction. The mechanism is the way to explain how you got there. The mechanism for granted. On exam day, on your own, with different structures, drawing mechanism is another story completely.
So what is the final (fast) step of the reaction? The second step above. Draw the arrow now. Electrophilic Addition: is named after the slow first step, formation of a higher energy species. If it's charged, it is higher in energy and thus more reactive. If you've drawn energy diagrams you know this. So how would you draw the arrows from here? What arrow is needed? Draw it in. You’ve started.
Arrows always move from what charge to what charge? From nucleophile to electrophile, from base to acid, essentially, always from negative species to positive species. What will the product be if you drew the correct arrow?
Did you obtain that answer? Hopefully you did and remember that the result of a reaction that occurs according to Markovnikov's Rule is regiochemically, is called the Markovnikov product. And it is major. Your professor doesn't care about that apparently. Had you given the anti-Markovnikov product (which would arise from the carbocation you were forbidden to draw when a more stable one is an option).
H-Br is a variable. It could be H-Cl, or H-I. HF is rarely used as fluoride is too reactive. The alkene is also a variable. Any number of different alkenes or alkynes will undergo this same sequence of arrows. Since the reagent and structure were given, good questions have one good answer. The arrows however are all the same. The hydrohalogenation reaction which alters one of the variables in the mechanism above.
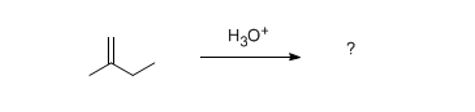
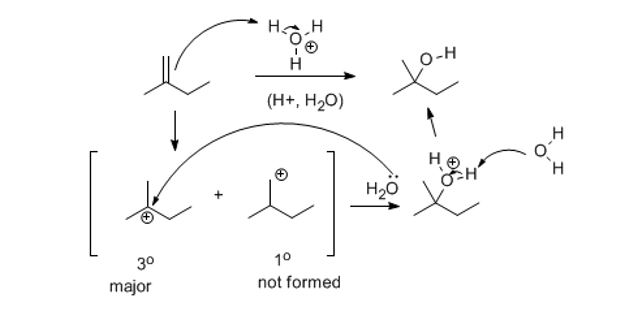
Hydration works the same way. Except it's acid-catalyzed which increases the number of steps by one. Try it please.
Stop here: Work the problem on your own now on separate paper.
Reason this out with me.
Answer the following: pause and think or discuss for 20 seconds about each one. So, as always, you have a nucleophile and an electrophile.
1) Which is the nucleophile?
2) What, by default, must be the electrophile?
3) What class of reaction is this? (not the name of the reaction, obviously hydration)
4) If these two species react, what will the potential organic intermediates look like?
5) Draw the major intermediate in brackets, as a step in the reaction.
6) What is this species called?
7) Now what two RELEVANT species exist in solution. (hint: one is the carbocation you made)?
8) The next step is another polar reaction. A reaction between two charged species (a nucleophile and an electrophile). Water is the nucleophile now and the carbocation must be the electrophile. If you have problems with identifying which is which here it’s ok, but if it becomes a persistent issue, we have major problems.
9) Draw the arrow that gets you closer to the product.
10) Draw what you get from that arrow.
In step 4, if you drew an arrow from the alkene to the formally + charged oxygen, you still don't get the point of polar covalent bonding and electronegativity. Sounds patronizing since that is general chemistry but it's not meant to be. If you drew it correctly then you understand the concept of formal charge (covered months ago at the beginning). If you didn't cover formal charge or are “iffy” about it, review it. |
__________________________________________________________________________________________________________________
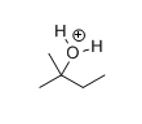
At this point, you should have the following structure:
If this is not what you have at this point,please go back now on separate paper and start over.
If this is where you are, draw arrows that remove the excess proton to give an alcohol (work-up).
That was one problem. A tough problem. In fact, for a beginning reaction, it doesn't get much harder (so sigh in relief).
NOTE:
This is how you should be studying every day. Every single day. If you're not, you are asking, no begging...to fail the class. Even if you only do one problem just like the one we did, that's OK.
If you see a new reaction, your first question should be, what is the mechanism? It doesn't just go whooosh! and it's over, does it?
Most want to pretend it does, and avoid drawing mechanisms for each problem. These people are always the same people who fail the class or withdraw. Or they barely pass with the curve, and go on to fail semester II miserably.
DRAW every day, working problems, starting now. If you already are, you wouldn't need me (or anyone else).
If you are just learning what mechanism is at this moment, make that change right now, or you will not pass this class. I can guarantee it, as I've seen a million times. If you are working too much at your job, you will have to manage your time more effectively.
Realize that if you haven't drawn the mechanisms (or realized you don't need to draw one) you are not doing anything. Maybe you've read the textbook, even cover to cover. That's not studying organic. If you don't know the mechanisms like the back of your hand, by working every problem you can and checking the answer with a solutions manual, you're going to struggle hard or fail (50% do every semester across the country) when it could be so much simpler. If you don't believe it, you haven't taken it. Your professor, your school’s prestige level etc. make a difference, but only a tiny one. All of this, is on you. It is ‘just you’ for everyone.
If he or she didn't assign any end of chapter problems, do them all yourself anyway. If you don't work problems until you know the RELEVANT mechanisms by heart, you will barely pass (you'd have to memorize everything (impossible, even if you have a photographic memory). It's like that for everyone.
How long did it take?
No one can escape this process and still get a decent grade for it (decent = B+ and above). Knowing it for next semester...you still need this for that too. It's cumulative until the end of the second semester not just for the next midterm exam.
Moving on: The Work-up
How did you draw the arrows to give the final neutral product?
If you haven't drawn those arrows, try it now on a separate sheet of paper. If you make mistakes that's fine, that's the only way to learn this. This is actually a large unspoken rule. Many who finish organic chemistry don't even know it is called a work-up when a reaction has gone to desirable conversion to product. If you start a reaction by adding strong acid to it, when you're done, what do you have to do to neutralize it? Add a base to remove the acid; and vice versa.
Here is the entire mechanism: if you didn't get this, rewrite the whole thing 5 times (I don't mean copy it down 5 times. I mean start from scratch, do it from the beginning, reason through it until you've attempted it 5 times or flawlessly, whichever comes first. (Without looking!) Do this for every problem on this exam or any reaction in your notes that s/he demonstrated in lecture. (also, a few reactions have no mechanism you need to know which is discussed below...those are for "note-card" making). Notice, this is not memorization. It's doing the same fundamental things repeatedly until you get the hang of it. Here's the complete mechanism:
There is one more 'subtlety' about Electrophilic Addition that you must learn. Bridged intermediates: specifically, the halonium and mercurinium ions. If you need to refer to previous pages or the textbook, do so.
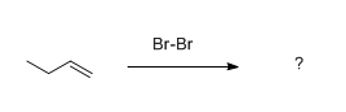
HALOGENATION (ADDITION OF X2): Br2, Cl2, I2
Just from what you were given, what will be the answer to this problem?
The overall outcome, that is.
It's obvious to anyone that both Br's will attach to each carbon of the double bond. There's nothing else present. But the intermediate during the reaction is different, not a carbocation. But a bridged intermediate (a three-membered-ring is more descriptive).
Next you will attack with the only nucleophile present which is..?
Where does it attack? If you don't know, find it right now in your textbook. Lookup cyclic halonium or bromonium ion or wiki it. Do not continue until you do.
Ready? Do you have a good idea what it is and how it appears?
It attacks at the more hindered carbon from the backside. (that's where the real positive charge is, don't confuse formal charge and electron density, they are different things)
Despite the fact, that this carbon is less accessible and both are partially positive, it is the one that is attacked. When you learn about transition states, you'll learn that there's an energy of activation. This barrier is lower depending on the imaginary transition state that forms. It's imaginary because bonds are forming and breaking. Which carbon you attack, will be a carbocation for a fraction of a second (nano- or femtoseconds). The answer is it wants the one that forms an almost carbocation at the secondary position not the primary. Here it doesn't make a difference because the electrophile added first, and the nucleophile added second is the same thing. As you attack the back side of this carbon...why the back side (that's where the dipole is positive). A bond must break, or you violate the octet rule. Thus the answer from the mechanism is:
If you think about it, most people draw the first step as shown above. Can you make two bonds with just one arrow? No, you can't. One arrow becomes one bond. One arrow=one bond. Draw the other arrow that is necessary to do this to make the ring. (hint: it comes from Br and goes where?)
Doesn't seem to matter what the regiochemistry is for halogenation...but it does if the solvent is water, which is known as the HALOHYDRIN reaction. In such a case, where would the alcohol group form? Water is solvent, therefore it can be regarded as being infinite in concentration (It's 55.5M) but that's infinite to a solution of reacting molecules in water. In other words, which is the correct answer to the following question: __________________________________________________________________________________________________
Draw all arrows, relevant lone pairs, formal charges, and each step to predict the product. If you can't do this, re-read the above. And start over if necessary.
___________________________________________________________
Another 3-membered ring intermediate of importance is the OXYMERCURATION/ DEMERCURATION reaction. It is crucial you know this as it reappears on every standardized exam involving organic chemistry. Same is true with HYDROBORATION / OXIDATION (see below).
Oxymercuration proceeds as exactly like the halohydrin reaction you did above: All the arrows are the same except pointing to different variables. In this case the electrophile is Hg or mercury.
What's the first step? Remember, this also forms a bridged or cyclic intermediate.
Now where does the water attack? Indicate with the proper formal charges.
What did you get as a result? Draw it
Was it:
Do this reaction from scratch, repeatedly, until you have memorized the mechanism. You should do this enough times, (vary the starting material) until you can do it on any alkene, for example. The arrows are exactly the same as the halohydrin reaction at the top of the page.
Ok, so now the demercuration...for which there is no mechanism you can draw. It's well known except way beyond the scope of this class. It's a redox reaction. We don't normally draw mechanisms for oxidation or reduction reactions. These are flash card reactions...do not even attempt to draw the mechanism for demercuration which is shown below. That is PhD level chemistry and is a complicated reaction with several kinetic equations.
You will lose points, in fact, if you attempt to draw such arrows as coming from borohydride to remove mercury. It is a radical reaction so even though some people draw a back sided attack of H- to the carbon with mercury on it, it is a giant oversimplification to ever do that. Know that sodium borohydride reduces the mercury, and a hydrogen takes its place. It just disappears. That's all you care about.
The product of oxymercuration or alkoxymercuration (if the solvent is an alcohol, instead of water) is a Markovnikov alcohol or ether? Doesn't acid-catalyzed hydration make the same thing? Why all the alphabet soup and toxic mercury?
Acid-catalyzed HYDRATION is an awful reaction that no sane organic chemist would ever actually do. Why not?
1) it forms racemic mixtures (mixtures of anything is inefficient and a waste of money) organic chemists go after the exact major product they desire, they don't just figure out mechanisms and solve questions.
2) it proceeds through a carbocation intermediate...problem:
CARBOCATIONS REARRANGE. (refer to your textbook, look up 1,2-hydride or 1,2-alkyl shift).
3) Both show up repeatedly either way, so know both and the realize the stupidity of doing acid-catalyzed hydration to make anything of value. You need to know stereochemistry first so we'll go there next.
Before that you need to know the following odd reactions (the first won a Nobel Prize).
Hydroboration-Oxidation is a famous reaction you must know forever (the end semester 2) that makes an anti-Markovnikov alcohol. For that reason, it won the Nobel prize and why you have to know the first step backwards and forwards. But not the second step (that's an oxidation).
This is how it proceeds. Since boron is a group III metalloid, it doesn't have a full octet when it's neutral. Therefore, it's the ficklest of the elements in the 2nd row. It wants a full octet, who doesn't? Beryllium and lithium When it gets one however, its formal charge is minus one. So it then wants to get rid of a bond to something that takes those electrons with it. In other words, it accepts, then loses, then accepts, then loses bonds to Lewis bases. It is a perfect atom for rearranging things. This is used to advantage in organometallic chemistry.
The first step is a concerted process. (all arrows move at the same time). Just like in halogenation (addition of Br2...right?)
Why does it attack boron, not the hydrogen? Look at the periodic table. What's more electronegative, H or B? Ok so, Boron is the electrophile, not hydrogen. So Markovnikov is still right. After the oxidation step (no mechanism you need to worry about, unless asked). You get an alcohol.
An anti-Markovnikov alcohol.
So far we have not violated Markovnikov's rule. What about the following?
Remember the addition of H-X and its regiochemistry? How could we get an anti-Markovnikov alkyl halide instead?
It's simple, add peroxide.
This is a radical reaction substitution not an electrophilic addition. That is another chapter. Just know for now (unless you did cover radicals already), that adding peroxide to the same reaction, switches the regiochemistry to anti-Markovnikov. Just know it.
Ready to conquer organic chemistry with confidence? Explore our services and resources now to start your journey towards success! Join our community of learners and unlock your full potential in organic chemistry. Let's embark on this exciting journey together. Get started today!